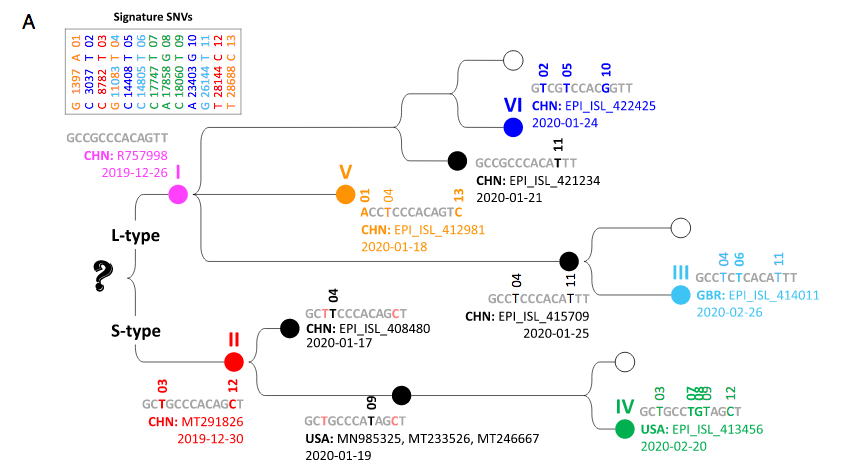
El síndrome respiratorio agudo evere coronavirus 2 (SARS-CoV-2), el agente causal del COVID 19, continúa evolucionando desde su primera aparición en diciembre de 2019. Usando las secuencias completas de 1.932 genomas del SARS-CoV-2, varios análisis de agrupamiento identificaron consistentemente seis tipos de cepas. Independientemente de la construcción del dendrograma, se identificaron 13 variaciones de firma en forma de variaciones de un solo nucleótido (SNV) en las regiones de codificación proteica y un SNV en la región 5 ‘no traducida (UTR) y proporcionaron una interpretación directa para los seis tipos (tipos I a VI). Los seis tipos de cepas y sus SNV de firma subyacentes se validaron en dos análisis posteriores de 6.228 y 38.248 genomas de SRAS-CoV-2 que estuvieron disponibles más tarde. Hasta la fecha, el tipo VI, caracterizado por los cuatro SNV de firma C241T (5’UTR), C3037T (nsp3F924F), C14408T (nsp12 P4715L) y A23403G (Spike D614G), con fuertes asociaciones alélicas, se ha convertido en el tipo dominante. Dado que C241T está en el 5’UTR con significado incierto y las características pueden ser capturadas por los otros tres SNV fuertemente asociados, nos enfocamos en los otros tres. La frecuencia cada vez mayor del haplotipo tipo VI 3037T-14408T-23403G en la mayoría de las muestras enviadas en varios países sugiere una posible ganancia de aptitud conferida por los SNV de firma de tipo VI. El hecho de que la falta de una o dos SNV de naturaleza siginosa no persista implica posibles interacciones entre estos SNV. Los SNV posteriores como G28881A, G28882A y G28883C han emergido con fuertes asociaciones alélicas, formando nuevos subtipos. Este estudio sugiere que las SNV pueden convertirse en una consideración importante en la clasificación y vigilancia de SARS-CoV-2.